Meet the Salis Lab. Learn how we use biophysical models and methods to rationally predict and control the behavior of biological organisms.

(Left to Right) Grace Vezeau, Alex Reis, Sean Halper, Daniel Cetnar
Grace Vezeau
Current sensing technologies for diagnostic compounds, like human biomarkers or environmental pollutants, rely upon expensive and non-robust equipment. My research seeks to solve this problem by developing RNA-based biosensors. Certain RNA sequences, called aptamers, fold into specific shapes and bind strongly and specifically to target ligands. We can couple this sensing element to a measurable output, such as the expression of a fluorescent protein, by designing the RNA molecule to change shape upon ligand binding. The RNA molecule acts as a switch, where output is turned on when the ligand is present.
I presented my work on TNT-detecting biosensors at the Fall 2015 SynBERC retreat, and will be presenting a broader overview of RNA-based sensors at the Rustbelt RNA meeting this October.
My favorite (set of) equations are the Lotka-Volterra equations, two differential equations describing the population dynamics of predators and their prey. They are one of the first examples of using mathematics to successfully describe a biological phenomenon – a tradition we are carrying through today.
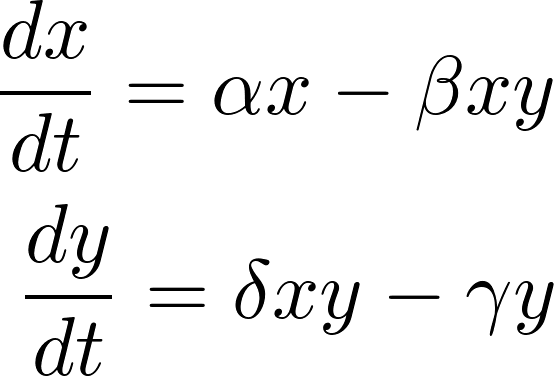
Alex Reis
Precise control over protein expression is required when engineering biological systems. My work is focused on developing sequence-to-function models of gene expression and regulation to allow programmable system behavior encoded at the DNA sequence level.
One of the most direct ways to encode the desired protein level in bacteria is to modify the ribosome binding site (RBS), a section of the protein’s mRNA that governs the rate at which the ribosome is recruited to initiate translation. The RBS Calculator is a thermodynamic equilibrium model used to predict relative protein level given an mRNA sequence (figure). Our group has steadily improved this model since 2009 (Salis et al., Nature Biotechnology), but we have identified subclasses of mRNA that fold slowly or incorrectly that are still poorly predicted. Inspired by these insights, I am building a non-equilibrium model of translation initiation that accounts for these RNA folding dynamics. To do this, I’ve accelerated an existing kinetic Monte Carlo method of RNA folding (FORTRAN) and will soon be building a Markov model to describe the transitions and interactions between an mRNA and the ribosome during translation initiation (Python).

I presented this work at the last Synberc retreat this past Spring at UC Berkeley. I’ll be giving a talk on this model comparison and analysis at AIChE this November in San Francisco (Bioengineering session).
As a side project, Sean, Phillip Clauer (undergrad), and I have also been working on the rational design of nonrepetitive sgRNAs for use in multiplex CRISPR/Cas9 applications. CRISPR is a powerful tool that now allows us to regulate and edit genes in a precise, targeted manner. One major shortcoming of this system is that you can only do one edit or gene knockdown at a time because you are limited by the expression of only one sgRNA. Our nonrepetitive sgRNAs will allow easy DNA synthesis and stable, robust expression of many sgRNAs. This will enable multiple simultaneous gene edits in genome engineering applications, multiple up- and down- regulations of genes in fundamental expression studies, and more complex logic than currently possible in synthetic genetic circuits.
On my mind a lot is the fluctuation theorem (restated as Crooks Fluctuation theorem) from statistical mechanics. The fluctuation theorem can give insights into the nonequilibrium behavior of biology (proteins, RNA, etc.) at the microscopic level. Stated in a general mathematical form below, as the time or system size increases, the forward time trajectory is exponentially more likely than the reverse, given that it produces entropy. In other words, there is always some nonzero probability that the entropy of an isolated system might spontaneously decrease. The second law of thermodynamics is a statistical one!

Sean Halper
In order to bring a desired biochemical product to market, companies and research groups often need to improve the titers of the pathway expressing their product of interest. However, the design-build-test cycle they use to maximize the productivity of pathways via the tuning of enzyme expression is too time-consuming and costly for pathways of sufficient size. My work with the Pathway Map Calculator uses de-dimensionalized kinetic models to model and predict the relationship between enzyme concentration and final product titer for a variety of pathways, while requiring less pathway variant data for large pathways than other methods.
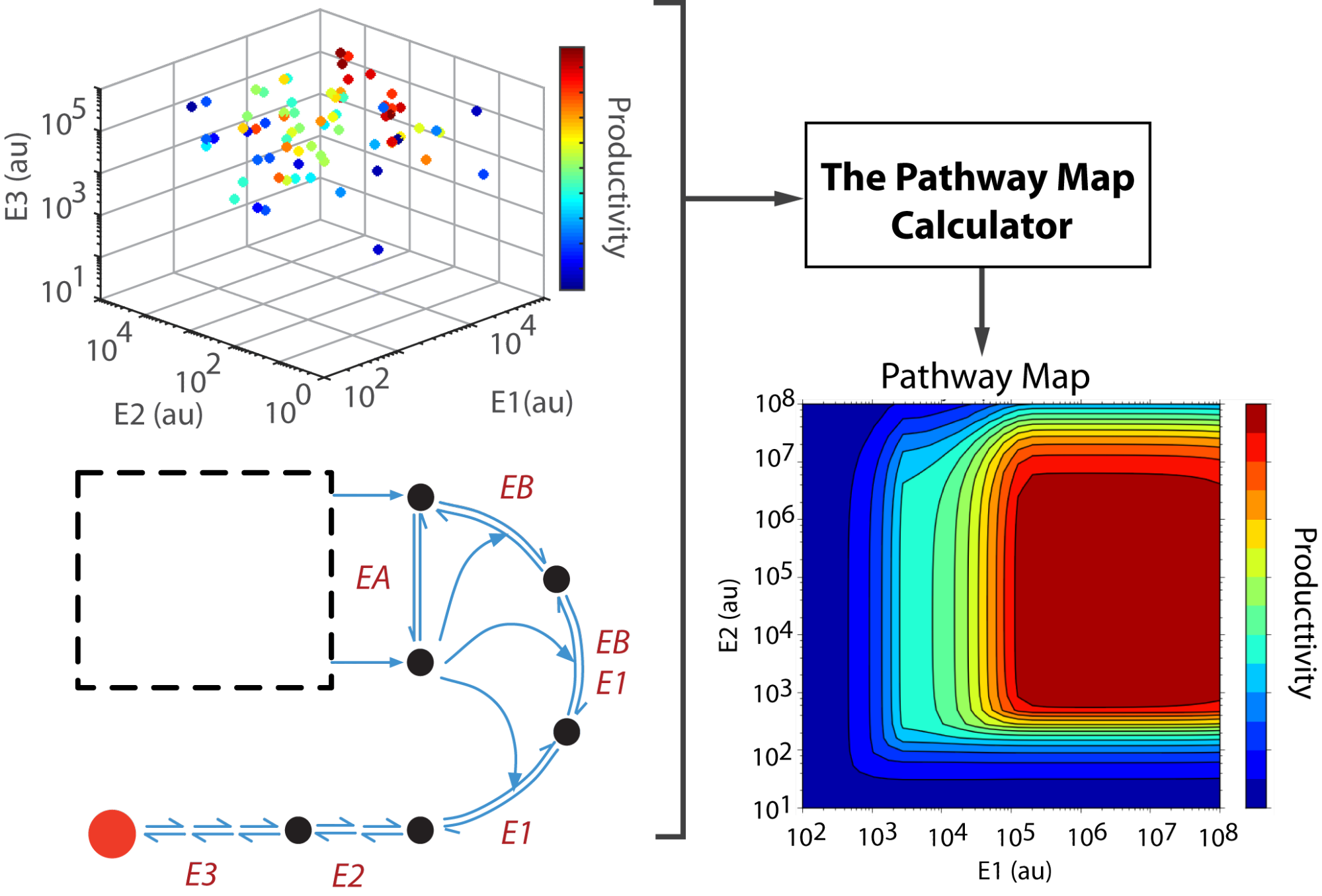
I have presented my work at the Fall 2015 meeting of Synberc in Boston, MA and will be giving a presentation at AIChE this November in San Francisco.
My favorite equation is the semi-official formulation of Murphy’s Law, where Pm is the probability of a social or mechanical malfunction; Km is Murphy’s constant (1); I, C, and U are the importance, complexity, and urgency associated with a given malfunction of frequency F on a base 10 subjective scale, respectively; and Fm is Murphy’s factor (approximately 0.01).
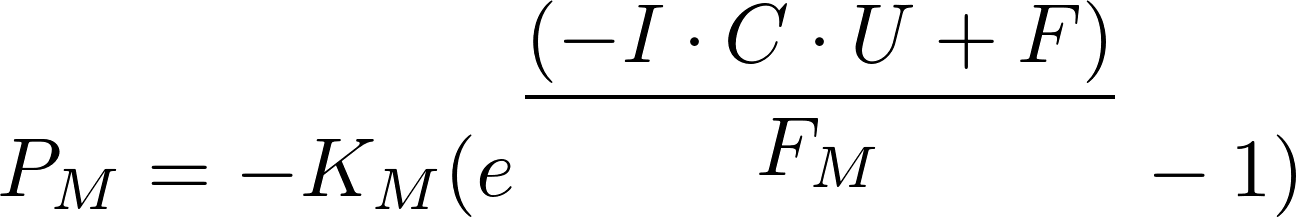
Daniel Cetnar
The use of natural products in medicine remains immensely important with active research occurring in anticancer, antibacterial, and antimicrobial drug development. Today, nearly 70% of all anti-infective drugs derive from natural products.
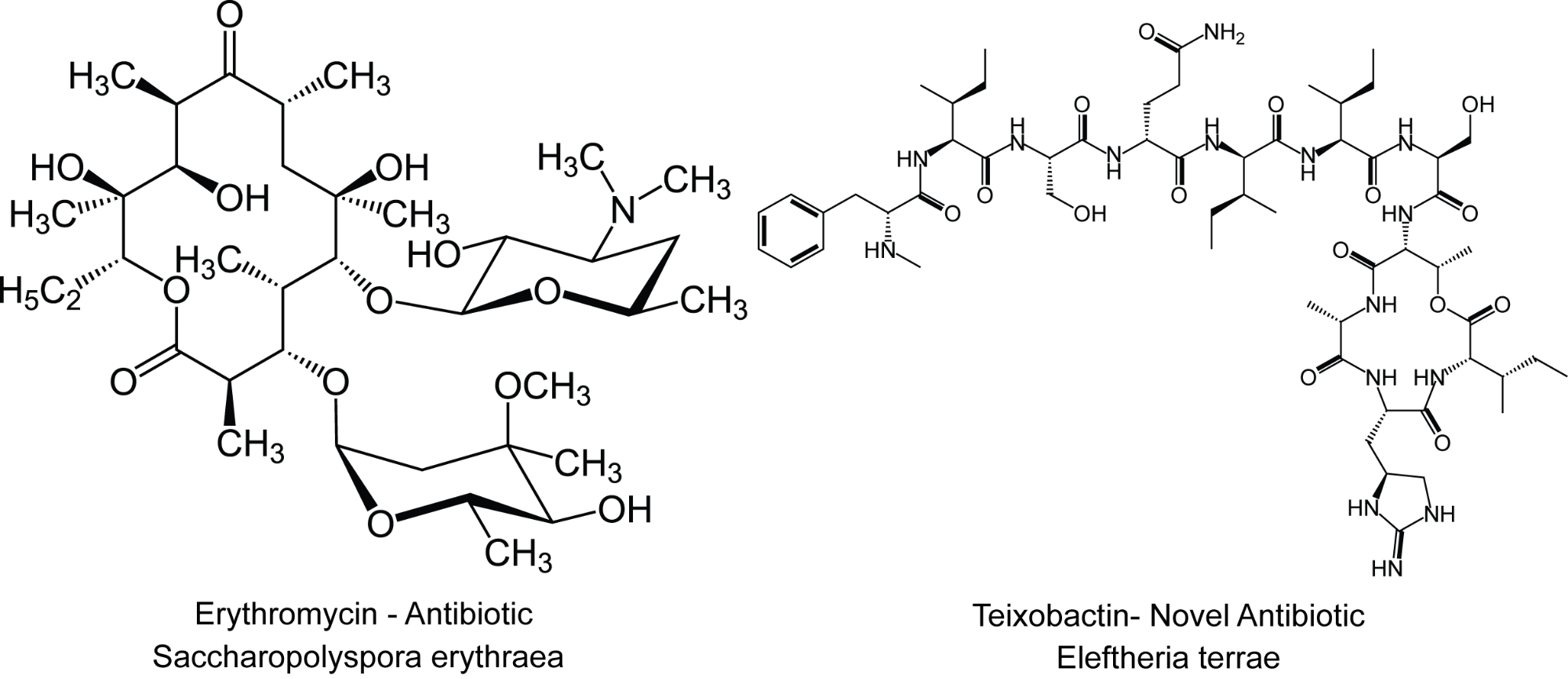
Interestingly, thousands of sequenced natural product gene clusters exist whose products have never been produced in appreciable quantities due to challenges in producing them in the native host. From sequence homology analysis, many of these natural products likely have useful properties, but have not been produced and confirmed. Discovering high-throughput methods to re-engineer sequenced gene clusters for efficient production in industrial bacterial strains remains immensely important for future drug development. My work aims to develop models and design rules to streamline gene cluster engineering for incorporation into the Operon Calculator. Currently, my work centers on understanding, quantifying, and modeling the structural basis of mRNA degradation to predict mRNA stability and design robust operons for reliable gene expression.
I presented my initial findings on mRNA degradation at the summer 2016 Synthetic Biology: Engineering, Evolution & Design (SEED) conference in Chicago, IL.
My favorite equation is the Hill equation because of its broad usefulness in describing biomolecular interactions. Many of the fundamental papers in Synthetic Biology make use of the Hill equation to describe genetic circuits and gene regulation.
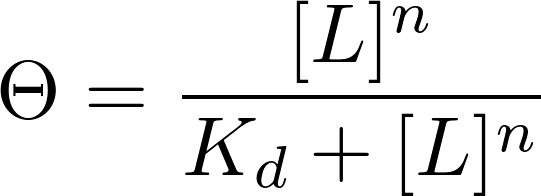
On behalf of the ChE GSA, we would like to thank students from the Salis lab for putting together this awesome summary.